BPHunter standalone is for users who want to run it locally in a command-line environment (also in GitHub).
Current Version: 2
Please download and replace the programs and reference datasets.
Programs:
BPHunter_VCF_one.py
BPHunter_VCF_batch.py
Dependency:
The code is written in python3, and requires bedtools installed.
Reference Datasets (GRCh38):
Download them and put them in the same folder with the BPHunter programs.
The following BED files could be loaded to IGV for visualizing BP locations, aligned with the variants of interest.
File Format:
Input: Variants in VCF format, with 5 mandatory and tab-delimited fields (CHROM, POS, ID, REF, ALT).
Output: BPHunter-detected variants will be output with the following annotations.
Command:
python BPHunter_VCF.py -i variants.vcf
python BPHunter_VCF.py -i variants.vcf -g GRCh37/GRCh38 -t all/canonical
arguments:
-h, --help show help message
-i, --input input variants in VCF-format file
-g, --genome human genome assembly {GRCh37, GRCh38}, default: GRCh37
-t, --transcript all/canonical transcript? {all, canonical}, default: all
python BPHunter_VCF_batch.py -d /dir -s samplelist.txt -o output.txt
python BPHunter_VCF_batch.py -d /dir -s samplelist.txt -o output.txt -g GRCh37/GRCh38 -t all/canonical
arguments:
-h, --help show help message
-d, --dir directory of VCF files
-s, --sample sample list (without .vcf extension) to be screened in the above directory
-o, --output output CSV filename, comma-delimited
-g, --genome human genome assembly {GRCh37, GRCh38}, default: GRCh37
-t, --transcript all/canonical transcript? {all, canonical}, default: all
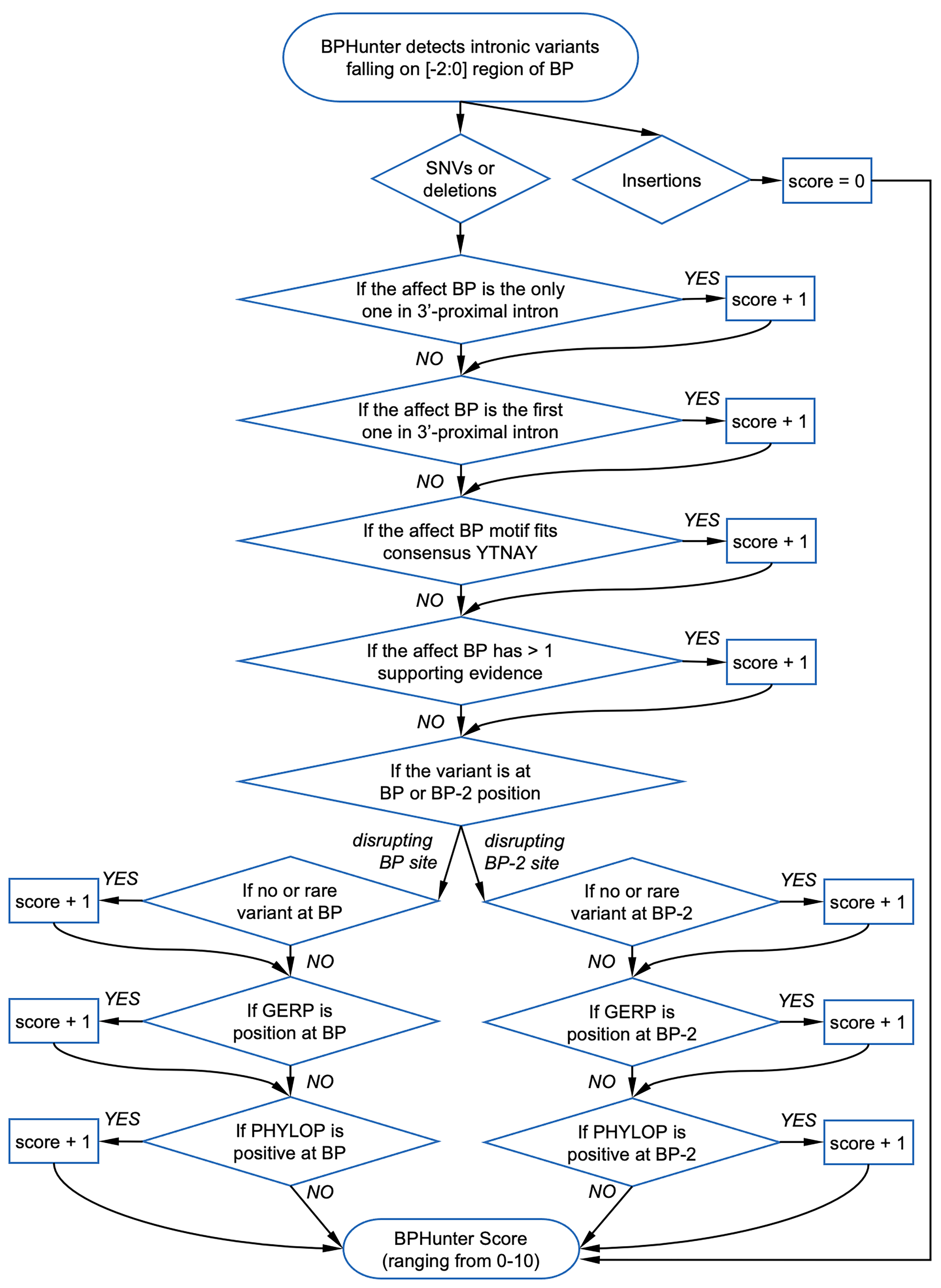
BPHunter Scoring Scheme:
We recommend to keep SNVs and deletions with BPHunter_HIGHRISK = 'YES', and then prioritize by BPHunter_SCORE.