AGAIN standalone is for users who want to run it locally in a command-line environment (also in GitHub).
Current Version: 1
Programs:
AGAIN_VCF_one.py
AGAIN_VCF_batch.py
Dependency:
The code is written in python3, and requires bedtools installed.
Reference Datasets (GRCh38):
Download them and put them in the same folder with the AGAIN programs.
File Format:
Input: Variants in VCF format, with 5 mandatory and tab-delimited fields (CHROM, POS, ID, REF, ALT).
Output: AGAIN-detected variants will be output with the following annotations.
Command:
python AGAIN_VCF.py -i variants.vcf
python AGAIN_VCF.py -g GRCh37 -t all -i variants.vcf
arguments:
-h, --help show help message
-g, --genome human genome assembly {GRCh37, GRCh38}, default: GRCh37
-t, --transcript all/canonical transcript? {all, canonical}, default: all
-i, --input input variants in VCF format file
python AGAIN_VCF_batch.py -d directory -s samplelist.txt -o output.txt
python AGAIN_VCF_batch.py -g GRCh37 -t all -d directory -s samplelist.txt -o output.txt
arguments:
-h, --help show help message
-g, --genome human genome assembly {GRCh37, GRCh38}, default: GRCh37
-t, --transcript all/canonical transcript? {all, canonical}, default: all
-d, --dir directory of VCF files
-s, --sample sample list (without .vcf extension) in the above directory
-o, --output output filename
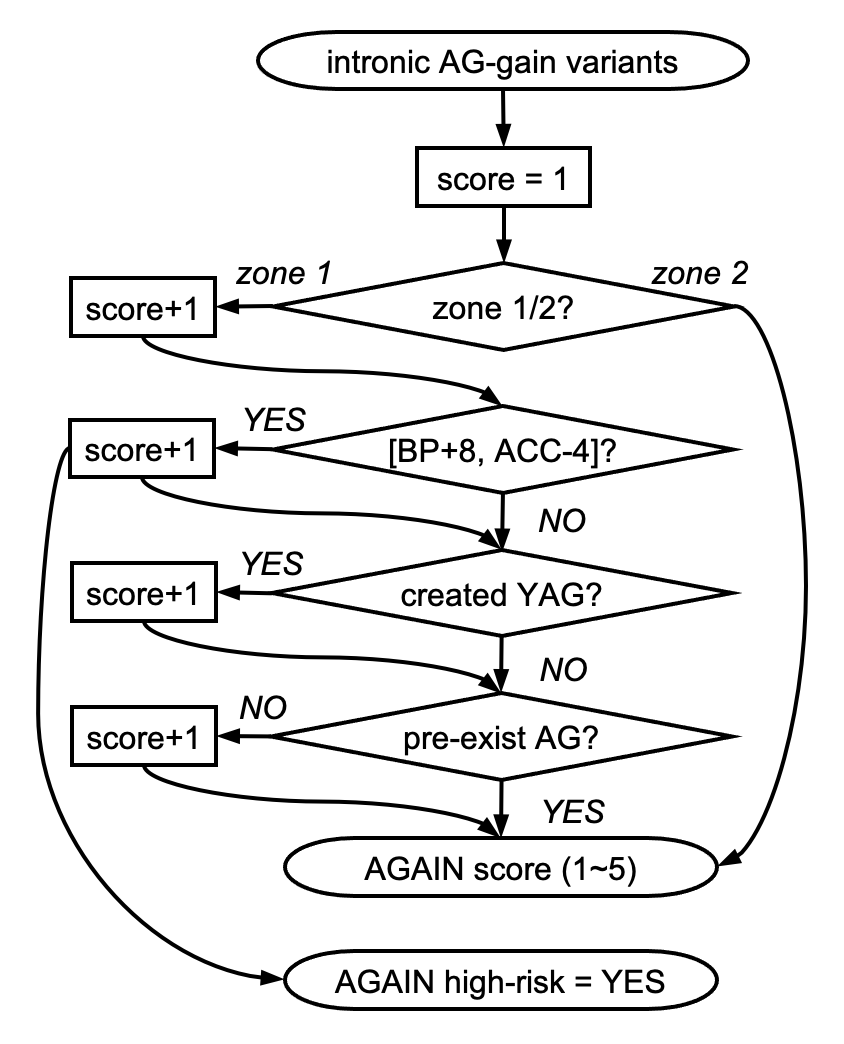
AGAIN Scoring Scheme:
We recommend to focus on the AG-gain variants with AGAIN_HIGHRISK = 'YES', and then prioritize by AGAIN_SCORE.